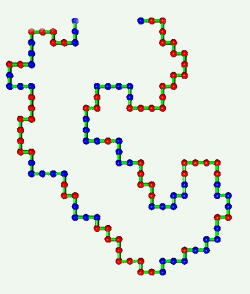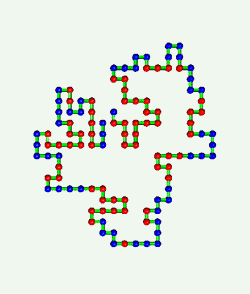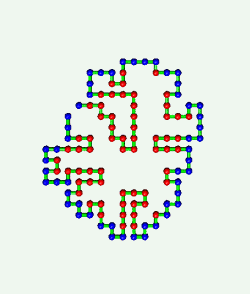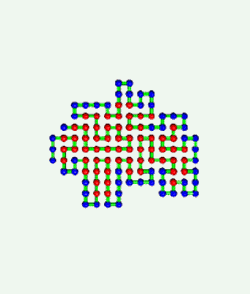
Proteins are born resembling strings of beads. Then, as shown in the
computer simulation at the left, they fold in an instant into intricate
patterns that make them become brain, blood, biceps, and bone.
ADVERTISEMENT (article continues below)
But
sometimes something goes awry in the folding process. One result is
disabling diseases like Alzheimer's and "mad cow" syndrome. They also
cause respiration and locomotion failures because protein functions
cannot be fully carried out. Yet, until now, biophysicists could only
guess at what happened.
The problem was that even supercomputers were not up to the
enormous computational task of describing the interactions between more
than one protein molecule. In fact, no one had devised a computer
algorithm that would model the interaction between just two protein
molecules until Jonathan King of Massachusetts Institute of Technology
recently teamed up with researcher Sorin Istrail of Sandia National
Laboratories' computational biology project to do so.
"What Sorin has done is to say, 'If bumping is important, I
will have a pair of strings move in space and bump into each other.' He
quickly discovered properties of folding that were absent in previous
simulations," says King, who pioneered "wet" laboratory experiments to
uncover protein-misfolding mechanisms. A paper describing the work will
be published this spring in the Journal of Computational Biology.
Using the model, developed by Istrail and MIT graduate student Russell
Schwartz, the researchers tracked two highly simplified proteins
interacting on a grid during millions of computerized trials. They
found it was the apparently random positioning of water-loving
(hydrophilic) molecules in a protein that prevents water-hating
molecules (hydrophobic,which possess the dominant folding force), from
binding with other proteins.
In Alzheimer's and other protein-based diseases, an amino acid from one
protein links to an amino acid on a second protein, rather than waiting
to link to one on its own chain. This protein "adultery," which occurs
as each protein passes through a series of intermediate folding steps,
results in proteins stuck to each other in inert masses.
The researchers found that, in an aqueous solution, amino acids
usually hook up with others in their own string because of the
randomness with which water-hating amino acid molecules are
interspersed among water-loving ones on the same protein. The
water-hating molecules are prevented from combining with their
counterparts on other strings by the action of water-loving molecules,
which form little pockets of protection around neighboring water-hating
molecules. These pockets require another protein to have exactly the
right shape to inject a corresponding amino acid into the tiny coastal
boundaries created by the protectors. Which rarely happens.
"Our Monte Carlo [statistical] simulations unveil how the
kinetic competition between folding and aggregation is resolved by the
'good' sequences' built-in protection against aggregation, due to their
hydrophobic-hydrophilic pattern," says Istrail. "This allows them to
escape misfolding traps and move towards the hypothesized 'fast track'
folding that takes place for real proteins".
The results will help laboratory scientists understand the
mechanisms by which incomplete folds occur, in order to prevent them.
"This is a step toward successful protein engineering," says Istrail.
"It provides our first clue in how to design sequences of laboratory
proteins that can survive the essential but complicated folding
process."